
Legal Framework Analysis
modRNA Vaccine Production in Clinical Trials vs. Mass Scale

The Public Readiness and Emergency Preparedness Act (PREP) provides immunity to covered entities against claims related to the administration of medical countermeasures during a public health emergency, except in cases of willful misconduct
The PREP Act and EUA liability shields are not absolute. Evidence of willful misconduct—including intentional use of SV40 promoters, fraudulent quality control, and reckless distribution of contaminated batches—provides grounds to negate immunity.
This paper covers the scope of PREP Act immunity & EUA.
PREP Act immunity is broad but not absolute.
~ the ‘Immunity’ applies unless there's willful misconduct, which is defined as an act or omission that is taken intentionally to achieve a wrongful purpose, knowing it would cause harm, or with reckless disregardthe EUA (Emergency Use Authorization) context.
~ under EUA, manufacturers have liability protection unless there's fraud or intentional violation of FDA requirements

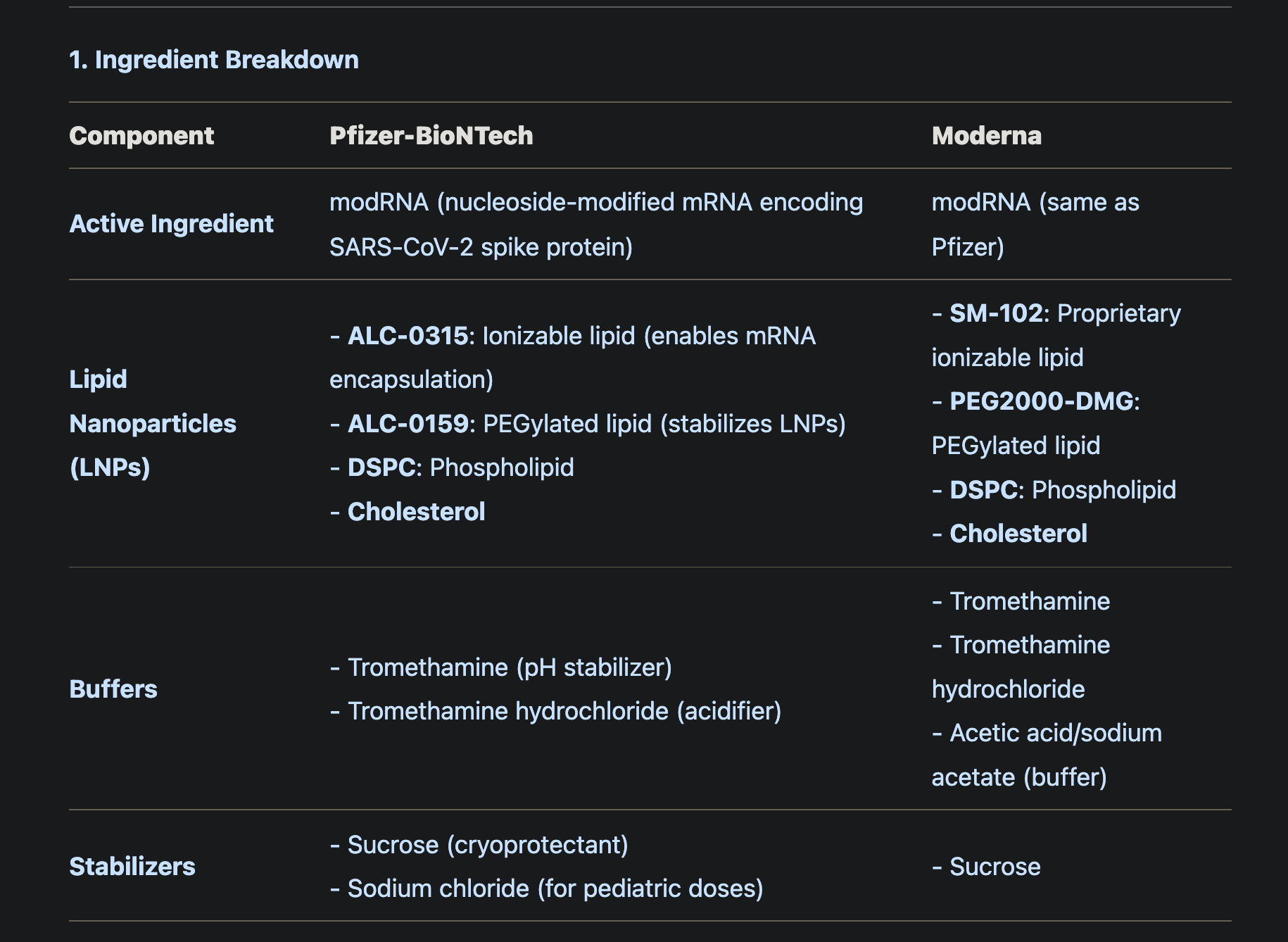
Propriatary Ingredients
Pfizer-BioNTech use ALC-0315 and ALC-0159 nanolipds
Moderna COVID-19 vaccines use SM-102 and PEG2000-DMG.
Ingredients
modRNA (Active Ingredient)
tromethamine (buffer)
sucrose (stabilizer)
Possible interactions: PEG-related allergies, lipid nanoparticles interacting with immune cells, buffer components affecting pH. Also, the presence of cholesterol in both could interact with cell membranes.
Potential Interactions and Safety Concerns
A. Lipid Nanoparticles (LNPs)
Ionizable Lipids (ALC-0315 vs. SM-102)
ALC-0315 (Pfizer): May transiently accumulate in liver/spleen, linked to rare cases of hepatitis in preclinical studies. Cleared within days.
SM-102 (Moderna): Controversy arose due to its use in chloroform (toxic in pure form), but vaccine-grade SM-102 is purified. No evidence of hepatotoxicity in humans.
Risk: Both lipids are transient and metabolized, but ionizable lipids may interact with cell membranes, triggering inflammation (e.g., myocarditis).
PEGylated Lipids (ALC-0159 vs. PEG2000-DMG)
PEG Allergy: Both vaccines use PEG derivatives. Pre-existing anti-PEG antibodies (common in ~7% of the population) increase anaphylaxis risk.
Autoimmunity: PEG’s structural similarity to human glycoproteins may theoretically trigger autoantibodies (no confirmed cases).
B. Residual DNA/Plasmid Contamination
Both vaccines use plasmid DNA (pDNA) in production. Residual pDNA fragments (e.g., SV40 promoters) could:
Integrate into host genome (theoretical risk).
Trigger TLR9-mediated inflammation (studies show low/no immune response to trace DNA).
C. Buffers and Stabilizers
Tromethamine (Both Vaccines)
Risk: May chelate calcium, potentially altering cardiac function (rare reports of arrhythmia).
Acetic Acid/Sodium Acetate (Moderna)
Risk: Low pH could irritate injection sites.
D. Sucrose/Sodium Chloride
Sucrose: Safe cryoprotectant; no known risks.
Sodium Chloride (Pfizer pediatric): Standard saline additive.
Unknown or Understudied Interactions
A. Lipid-Protein Interactions
LNPs may adsorb serum proteins (e.g., apolipoproteins), altering biodistribution. Moderna’s SM-102 LNPs show higher liver uptake vs. Pfizer’s ALC-0315.
B. Synergistic Toxicity
PEG + Ionizable Lipids: Combined effects on immune activation (e.g., complement activation) could amplify inflammation.
Residual DNA + LNPs: DNA fragments bound to LNPs may enhance nuclear uptake (SV40 promoter risk).
C. Batch Variability
Pfizer’s pediatric doses (with NaCl) showed higher rates of fever/seizures vs. adult formulations, suggesting ingredient interactions.

Regulatory Gaps and Unknowns
Long-Term Lipid Accumulation
Preclinical studies show LNPs cleared within weeks, but no human data beyond 2 years.
Genomic Integration
No evidence of DNA integration, but SV40 promoter bioactivity remains understudied.
Pediatric Formulations
Sodium chloride in Pfizer’s pediatric doses may interact with immature renal systems (unconfirmed).
Legal Implications
Failure to Disclose Risks: Neither Pfizer nor Moderna disclosed residual DNA or SV40 promoter risks in EUA filings.
Ingredient Variability: Batch-dependent adverse events (e.g., myocarditis clusters) suggest inconsistent LNP/mRNA ratios.
Key Issues
Transition from clinical trial production to mass production
The clinical trials used a cleaner, controlled process,
mass production involved E. coli and plasmid DNA with SV40 promoters.
Contaminants like plasmids and physical particles (metal, glass) were issues in mass production.
FDA's inability to inspect plants
Fraud: fraudulent quality control data
Falsified Western Blots and trial data misrepresented efficacy/safety 1
Reliance on Flawed Data:
FDA/EMA Fast-Tracking:
Pfizer’s EUA relied on Ventavia’s falsified trial data, yet the FDA granted full approval in August 2021 without re-inspection 2
EMA accepted low mRNA integrity batches, ignoring leaked quality control warnings
Negligence: Manufacturers failed to adhere to GMP and trial protocols
Failure to disclose risks (SV40 promoters, plasmid DNA) 3
Plasmid DNA Contamination:
Residual SV40 promoters in commercial batches risk genomic integration and indefinite spike production
Higher plasmid levels correlated with SAEs in Danish data
ignoring known manufacturing issues (contaminants, inconsistent batches)
Strict Liability:
Defective batches caused foreseeable harm e.g., myocarditis, clotting4
Batch variability & batch-dependent safety signals
Pfizer's leaked EMA documents showed low mRNA integrity in early batches.
Denmark’s SAE signals were dismissed by Germany (PEI) and the UK (MHRA), citing “methodological flaws
Moderna had issues too, like stainless steel contaminants in Japan
Moderna’s steel-contaminated batches in Japan were recalled, but similar incidents in the EU/US were not disclosed
►Legal Claim: Failure to act on SAE signals violates 21 CFR § 600.80 (post-marketing reporting) and constitutes reckless disregard under the PREP Act.
Whistleblower testimony
Brook Jackson of Ventavia Research;
unblinded participants
poor training of vaccinators
failure to follow up on adverse events
falsified audits:
Ventavia's practices were problematic, and Pfizer continued to use them for other trials even after the issues were reported.
Pfizer-BioNTech COVID-19 Vaccine contains: messenger ribonucleic acid (mRNA), lipids (((4-hydroxybutyl)azanediyl) bis(hexane-6,1-diyl) bis(2- hexyldecanoate), 2 [(polyethylene glycol)-2000]-N, N-ditetradecylacetamide, 1,2-distearoyl-sn-glycero-3-phosphocholine, and cholesterol), tromethamine, tromethamine hydrochloride, and sucrose.
Pfizer-BioNTech COVID-19 Vaccine for usein individuals 6 months through 4 years of age also contains sodium chloride
SPIKEVAX (COVID-19 Vaccine, mRNA), Moderna COVID-19 Vaccine, and Moderna COVID-19 Vaccine, Bivalent contain the following ingredients: messenger ribonucleic acid (mRNA), lipids (SM-102, polyethylene glycol [PEG] 2000 dimyristoyl glycerol [DMG], cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphocholine [DSPC]), tromethamine, tromethamine hydrochloride, acetic acid, sodium acetate trihydrate, and sucrose.
I. Inaccurate Clinical Trials and Falsified Audits
A. Pfizer-BioNTech Clinical Trial Misconduct
Whistleblower Evidence:
Brook Jackson, former Ventavia Research Group director, exposed falsified data, unblinded participants, and neglected adverse event follow-ups during Pfizer’s Phase III trials 1911.
Internal documents revealed:
Mislabeled lab specimens.
Vaccines stored at improper temperatures.
Staff instructed to falsify e-diaries and ignore protocol deviations 111.
Regulatory Failure: The FDA inspected only 9 of 153 trial sites, none from Ventavia, despite Jackson’s September 2020 complain
B. Moderna’s Trial Oversight Gaps
Lack of Transparency:
Moderna’s trials relied on proprietary LNPs and pseudouridine-modified mRNA, but independent audits of batch consistency were not disclosed
EMA leaks revealed mRNA integrity as low as 55% in commercial batches, raising questions about trial-to-production parity
Legal Claim: Fraudulent trial data submission violates 18 U.S. Code § 1001 and invalidates EUA prerequisites under 21 U.S. Code § 360bbb-3
II. Manufacturing Process Differences
A. Clinical Trial Production (Process 1)
Method: Small-scale, controlled enzymatic synthesis (e.g., in vitro transcription) with minimal contaminants.
Key Features:
High mRNA integrity (~100% intact).
Rigorous purification to remove endotoxins, dsRNA, and bacterial DNA.
No plasmid DNA or SV40 promoter sequences.
B. Mass Production (Process 2)
Method: Plasmid DNA (pDNA) amplification in E. coli bacteria with SV40 promoters.
Key Risks:
Residual pDNA: Incomplete removal of bacterial DNA during purification, confirmed by regulatory documents.
SV40 Promoters: Sequences from simian virus 40 (SV40) linked to nuclear translocation and genomic integration.
Antibiotic Resistance Genes: Potential horizontal gene transfer to human microbiome.
Legal Claim: Transitioning to Process 2 introduced known contaminants (e.g., plasmids, SV40) without adequate safety testing or disclosure, violating 21 CFR § 610.13 (freedom from extraneous material).
III. Contaminants and Safety Risks
A. Plasmid DNA and SV40 Promoters
Evidence:
FDA/EMA documents confirm residual pDNA in commercial batches.
Danish study found batches with higher plasmid DNA levels correlated with increased adverse events.
Risks:
Genomic Integration: SV40 promoters enable nuclear uptake, risking indefinite spike protein production or oncogenesis.
Microbiome Alteration: Residual antibiotic resistance genes could spread to gut bacteria.
Legal Claim: Failure to disclose plasmid/SV40 risks violates informed consent principles and 21 CFR § 201.57 (labeling requirements).
B. Physical Contaminants
Evidence:
Japan recalled 1.63M Moderna doses (2021) due to stainless steel particles.
Ryan Cole reported glass shards and inconsistent LNP/mRNA content in vials.
Visible contaminants indicate violations of Good Manufacturing Practices (GMP) under 21 CFR § 211.
III. Quality Control Failures
A. Inconsistent Formulation
Evidence:
Independent analyses (e.g., Del Bigtree/Ryan Cole) found batch variability: some vials lacked mRNA or LNPs.
Mass spectrometry of 4 vials detected LNPs but no mRNA.
Implications:
Suggests poor mixing, placebo substitution, or mRNA shortages, violating 21 CFR § 211.110 (sampling and testing).
B. Fraudulent Quality Control Data
Evidence:
Western Blots (intended to confirm antigen production) were allegedly computer-generated or showed unintended proteins.
Example: Fragmented mRNA producing non-target proteins.
Fraudulent data submission violates 18 U.S. Code § 1001 (false statements to regulators).
IV. Regulatory Negligence
A. Lax Oversight
Evidence:
Vanity Fair (2020) reported FDA’s inability to inspect manufacturing plants pre-rollout.
EMA leaks revealed acceptance of low mRNA integrity (55%) in commercial batches.
Failure to enforce GMPs under 21 U.S. Code § 331 (prohibited acts).
B. Suppression of Safety Data
Evidence:
BMJ investigations found regulators ignored Process 1 vs. Process 2 safety comparisons.
Whistleblower reports of clinical trial fraud (e.g., Brook Jackson, Ventavia) were dismissed.
Violation of Public Health Service Act § 402 (transparency in biologics regulation).
V. Legal Theories and Remedies
A. Product Liability
Strict Liability: Defective design (SV40 promoters) and manufacturing (contaminants).
Negligence: Failure to meet GMPs or disclose risks.
B. Fraud
Fraudulent Misrepresentation: False claims of safety/efficacy via falsified Western Blots.
RICO Claims: Coordinated suppression of adverse event data by manufacturers/regulators.
C. Regulatory Accountability
Mandamus Action: Compel FDA/EMA to release withheld safety data.
Class Action: For injured parties due to inconsistent/contaminated batches.
VI. Supporting Evidence
Documentation:
EMA leaks on mRNA integrity (PMC8787697).
Japan’s Moderna recall notices (2021).
Vanity Fair’s FDA investigation (2020).
Expert Testimony:
Toxicologists on plasmid DNA risks.
Biochemists on SV40 promoter bioactivity.
Regulatory Accountability
Injunctive Relief:
Halt distribution pending independent batch audits.
Congressional Investigation:
Subpoena FDA/EMA communications on ignored SAE signals
The Pfizer-BioNTech COVID-19 vaccine completed Phase 3 trials in November 2020, before being authorized for emergency use by the U.S. Food and Drug Administration. The study protocol runs until January 2023 because it includes a Phase 4 study, which is part of the post-marketing surveillance and not required for vaccine authorization or approval.
FDA | In December 2020, the Pfizer-BioNTech COVID-19 vaccine became the first COVID-19 vaccine authorized for emergency use in the U.S. Since then, the country has administered more than 202 million doses of this vaccine to its population. On 23 August 2021, the Pfizer-BioNTech COVID-19 vaccine reached a new milestone by receiving the first full approval of a COVID-19 vaccine in the U.S. This vaccine, which will be now marketed as Comirnaty, is approved for people aged 16 and older but remains under Emergency Use Authorization (EUA) for people aged 12 to 15 and as boosters.
Phase 3 trial of the Pfizer-BioNTech COVID-19 vaccine began in July 2020 and completed enrollment in January 2021. The trial involved 46,331 participants from 153 clinical trials sites in the U.S., Germany, Turkey, South Africa, Brazil, and Argentina. Phase 3 trial in adults was completed in November 2020, when Pfizer reported in a press release that the COVID-19 vaccine “met all of the study’s primary efficacy endpoints”. The results showing that the vaccine was 95% effective in preventing COVID-19 were published in the New England Journal of Medicine in December 2020
FDA NEWS 11/05/2021 | Pfizer and research partner falsified COVID-19 vaccine trials data | https://fda.news/2021-11-05-pfizer-falsified-covid-vax-trials-data.html
Former regional director (Brook Jackson, a trained clinical trial auditor with more than 15 years of experience in clinical research coordination and management) for Ventavia Research Group of Texas provided evidence that her former employer has falsified data, unblinded trial participants and neglected the follow-up of subjects experiencing adverse events when they conducted a Pfizer Wuhan coronavirus (COVID-19) vaccine trial in 2020.
UK MHR Rsponse
1) Is the MHRA aware of DNA contamination present in the mRNA COVID vaccines?
MHRA | We are not aware of DNA contamination in the mRNA COVID-19 vaccines, contamination as a term tends to relate to external material. Residual DNA which is considered as a process- related impurity is tightly controlled in all authorised Pfizer vaccines (Comirnaty). The purification and quality control process ensures that leftover DNA is within acceptable regulatory limits
2) Is the MHRA aware of the SV40 promoter sequence being present as a contaminant?
MHRA | No safety concerns related to residual DNA in the vaccine have been identified for any of the authorised vaccines
The Pfizer-BioNTech COVID-19 vaccine does not contain simian virus 40 (SV40). The presence of the SV40 promoter enhancer sequence is not the same as the presence of the whole virus itself. The SV40 promoter enhancer sequence was found to be a residual DNA fragment in Pfizer-BioNTech COVID-19 vaccine. The fragment is inactive, has no functional role, and was measured to be consistently below the limit required by regulators.
3) Is the MHRA aware the DNA is protected within the LNPs and as such will transfect human cells?
MHRA | We are aware that the residual DNA is could potentially be encapsulated within the LNPs, however, we do not agree that residual DNA in the LNPs will transfect human cells.
We are also not aware of any scientific evidence showing that the small amounts of residual DNA that may be present in the vaccine, could transfect into cells and integrate into the DNA of a vaccinated person. We are not aware of any scientific evidence showing that the small amounts of residual DNA that may be present in the vaccine could be a potential factor for cancer promotion.”
4) Has the MHRA ever independently tested the Pfizer/BioNTech vaccine at
the vial level?
MHRA | In the context of fragmented DNA and SV40 enhancer MHRA has not tested the Pfizer/BioNTech vaccine at the vial level.
Independent laboratory testing of vaccines is carried out by the MHRA’s
Official Medicines Control Laboratory (OMCL) (with a NIBSC certificate
being applied to compliant batches). The independent testing does not
verify the composition of the vaccine, rather it assesses key parameters
that focus on biological quality of the product. Independent assessment
also confirms that the manufacturer has reported on its wide-ranging tests
on the product. Batches of vaccine that meet the specifications in the
approval are certificated allowing the manufacturer to market them in the
UK for use before the batch expiry date.
In terms of the Pfizer/BioNTech vaccines tests conducted at the MHRA include:
Potency/sequence ratio, identity, RNA encapsulation, RNA content, RNA integrity.
All vaccine manufacturers must operate to Good Manufacturing Practices and
their facilities are licensed, and are inspected periodically. These procedures help to ensure that no batches of vaccine that may be contaminated get released in the UK.
5) Does the MHRA intend to test the Pfizer-BioNTech COVID-19 mRNA vaccine
for the presence of fragmented DNA and SV40 enhancer?"
MHRA | There are currently no intentions to test the Pfizer-BioNTech COVID-19
mRNA vaccine for the presence of fragmented DNA and SV40 enhancer.
We trust that you will find this information of use.
Reports of Batch-Dependent Suspected Adverse Events of the BNT162b2 mRNA COVID-19 Vaccine: Comparison of Results from Denmark and Sweden

Results: Significant batch-dependent heterogeneity was found in the number of SAEs per 1000 doses for both countries, with batches associated with high SAE rates detected in the early phase of the vaccination campaign and positive correlations observed between the two countries for the severity of SAEs from vaccine batches that they shared. Mild SAEs predominated in the batches used in the early part of the vaccination roll-out, where markedly higher SAE rates per 1000 doses in Denmark for the batches that were shared between the two countries suggested that a large proportion of these SAEs were under-reported in Sweden.
Three distinct clusters of BNT162b2 vaccine batches were identified, with highly variable SAE reporting rates. Also, a temporal reduction in batch-dependent SAE rates was found that paralleled the temporal sequence of vaccine batch roll-out, with batches administered at the beginning of the vaccination campaign displaying disproportionally high SAE rate
Conclusions: The batch-dependent safety signal observed in Denmark and now confirmed in Sweden suggests that early commercial batches of BNT162b2 may have differed from those used later on,
Additional Sources:
Schmeling M., Manniche V., Hansen P.R. Batch-dependent safety of the BNT162b2 mRNA COVID-19 vaccine. Eur. J. Clin. Investig. 2023;53:e13998. doi: 10.1111/eci.13998
Schmeling M., Manniche V., Hansen P.R. Batch-dependent safety of the BNT162b2 mRNA COVID-19 vaccine. Eur. J. Clin. Investig. 2023;53:e14102. doi: 10.1111/eci.14102
Christensen T., Jensen M.D., Kluth M., Kristinsson G.H., Lynggaard K., Lægreid P., Niemikari R., Pierre J., Raunio T., Adolf Skúlason G. The Nordic governments’ responses to the COVID-19 pandemic: A comparative study of variation in governance arrangements and regulatory instruments. Regul. Gov. 2023;17:658–676. doi: 10.1111/rego.12497